.svg)
%20(1).svg)

Manufacturing excellence in the medical device industry demands more than just innovative products, it requires a robust foundation of properly structured documentation that transforms compliance from a burden into a competitive advantage. When you master ISO 13485 requirements, you unlock the ability to scale operations while maintaining the highest standards of quality and regulatory adherence.
The ISO 13485:2016 Standards represent the global benchmark for medical device quality management systems, establishing clear pathways for manufacturers to demonstrate their commitment to patient safety and operational excellence. Understanding how to structure your medical device file ISO 13485 creates the backbone for sustainable growth in regulated markets worldwide.
What Constitutes a Medical Device File Under ISO 13485 Standard
The ISO 13485 standard defines a medical device file as a comprehensive collection of documents that demonstrates compliance with applicable regulatory requirements throughout a device's lifecycle. This file serves as the central repository containing all evidence necessary to prove your device meets safety, efficacy, and quality specifications.
BPRHub's Unified Compliance Framework centralizes these documentation requirements, ensuring manufacturers maintain audit-ready files across 30+ Standards including ISO, FDA, and GMP requirements. Rather than managing scattered documents across multiple systems, we create a single source of truth for your medical device file ISO 13485 compliance.
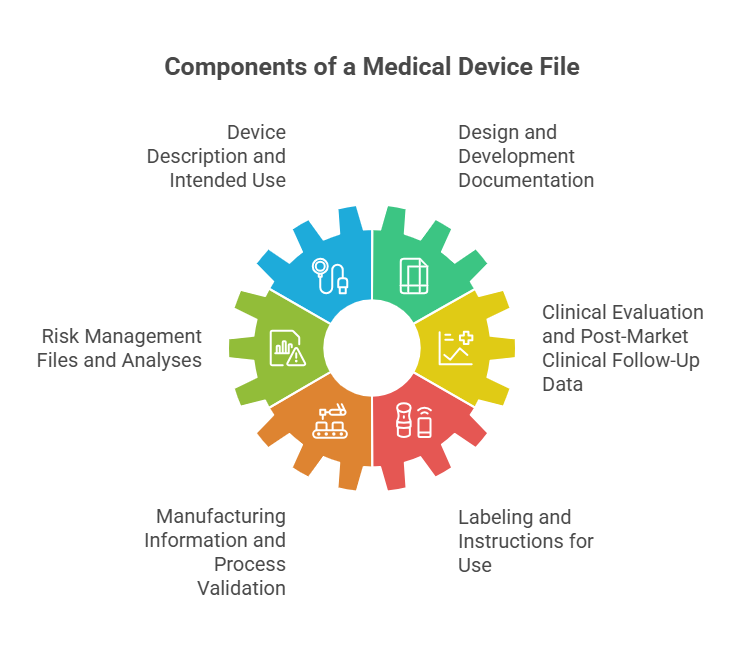
Core Components of Technical File Medical Device Documentation
Your technical file for a medical device must contain specific elements that demonstrate a a comprehensive understanding of your device's design, manufacturing, and intended use. The Standards require detailed documentation covering device specifications, risk management analyses, clinical evaluation data, and post-market surveillance information.
These components work together to create a complete picture of your device's safety profile and performance characteristics. Each element must be traceable to specific regulatory requirements and maintained throughout the device's commercial lifecycle.
Key technical file components include
- Device description and intended use specifications
- Design and development documentation
- Risk management files and analyses
- Clinical evaluation and post-market clinical follow-up data
- Manufacturing information and process validation
- Labeling and instructions for use
Medical Device File ISO 13485 Versus Other Regulatory Files
While the medical device file ISO 13485 shares similarities with other regulatory submissions, it serves a distinct purpose within your quality management system. Unlike FDA submissions that focus on market authorization, the ISO file demonstrates ongoing compliance with quality system requirements.
The ISO file integrates seamlessly with other regulatory documentation, while maintaining its unique focus on the effectiveness of the quality management system. This integration creates efficiencies that reduce duplication while ensuring comprehensive coverage of all regulatory requirements.
Read more about- Understanding Quality Management Systems for Medical Devices
What are the essential ISO 13485 Documentation Requirements for Device Files?
The ISO 13485 documentation requirements establish a structured approach to organizing and maintaining your medical device files. These requirements ensure consistency across your organization while providing clear guidance for document control and version management.
Effective documentation structures support both internal operations and external audits, creating transparency that builds confidence with regulatory bodies and business partners. When properly implemented, these requirements transform documentation from a compliance obligation into a strategic business asset.
Quality Management System Documentation Structure
Your medical device quality management system documentation must follow a hierarchical structure that clearly defines policies, procedures, and work instructions. This structure ensures personnel understand their roles while providing auditors with clear pathways to verify compliance.
The documentation hierarchy typically includes:
- Quality manual defining your QMS scope and approach
- Standard operating procedures detailing specific processes
- Work instructions provide step-by-step guidance
- Forms and templates supporting consistent execution
- Records demonstrating implementation effectiveness
BPRHub's centralized documentation system maintains this hierarchy while enabling real-time collaboration across teams. We ensure that version control and change management processes align with ISO 13485:2016 requirements.
Stay audit-ready—start your BPRHub trial today
📍 Book a Demo
📧 hello@bprhub.com
QMS Medical Device File Organization Standards
Organizing your QMS medical device files requires careful attention to logical grouping and cross-referencing capabilities. The Standards emphasize the importance of maintaining relationships between different document types while ensuring easy retrieval during audits or investigations.
Effective organization systems support both day-to-day operations and regulatory inspections. When auditors can quickly locate relevant information, it demonstrates the maturity of your quality management system and builds confidence in your compliance approach.
What are the Medical Device Requirements for File Content and Structure
Understanding medical device requirements for file content helps manufacturers avoid common documentation gaps that can lead to regulatory delays or compliance issues. The Standards specify minimum content requirements while allowing flexibility in how manufacturers organize and present information.
Content requirements vary based on device classification, intended use, and applicable regulatory pathways. However, all files must demonstrate a systematic approach to managing device-related risks and ensuring consistent product quality.
Device Master Record Integration with Medical Device Files
The device master record serves as the foundation for manufacturing operations while supporting broader compliance objectives through integration with your medical device file. This integration ensures manufacturing specifications align with design requirements and regulatory commitments.
Effective integration creates traceability between design inputs and manufacturing outputs, supporting both quality assurance and regulatory compliance objectives. When changes occur, integrated systems automatically update related documentation to maintain consistency.
Integration benefits include
- Automatic updates when design changes occur
- Clear traceability between specifications and manufacturing
- Reduced risk of documentation inconsistencies
- Streamlined change control processes
Design History File Medical Device Documentation Links
Your design history file medical device documentation must link directly to your ISO 13485 file to demonstrate compliance with design control requirements. These links create audit trails that prove systematic development approaches and regulatory compliance.
Design history files document the evolution of your device from concept to commercial product, while ISO files demonstrate ongoing quality management throughout the product lifecycle. Together, they provide comprehensive evidence of your commitment to patient safety and product quality.
What are ISO 13485 Medical Devices File Maintenance and Updates
Maintaining current ISO 13485 medical devices files requires systematic approaches to change control and version management. The Standards emphasize the importance of keeping documentation current while maintaining historical records for traceability purposes.
Change control processes must balance the need for current information with the requirement to maintain audit trails. Effective systems enable rapid updates while preserving the integrity of historical records.
Medical Device Documentation Control Procedures
Medical device documentation control procedures establish clear protocols for creating, reviewing, approving, and distributing documents throughout your organization. These procedures ensure consistency while supporting regulatory requirements for document management.
Effective control procedures include:
- Clear approval authorities for different document types
- Review cycles that ensure currency and accuracy
- Distribution controls that prevent use of obsolete versions
- Archive systems that maintain historical records
- Training requirements for personnel using controlled documents
QMS Documentation Version Control Systems
QMS documentation version control systems track changes while maintaining the integrity of your quality management system. These systems must support both regulatory requirements and operational efficiency needs.
Modern version control systems integrate with broader quality management platforms to provide real-time visibility into document status and change history. This integration supports both compliance objectives and operational efficiency goals.
What is ISO 13485 Compliance Through Proper Medical Device Files
Achieving ISO 13485 compliance requires more than just creating the right documents; it demands systematic implementation of processes that ensure ongoing effectiveness. Your medical device files serve as evidence of this systematic approach while supporting continuous improvement objectives.
Compliance is not a one-time achievement but an ongoing commitment to excellence that requires regular review and updating of your documentation systems. When properly maintained, medical device files become strategic assets that support business growth and market expansion.
Medical Device Validation Documentation Requirements
Medical device validation documentation must demonstrate that your processes consistently produce products meeting predetermined specifications. This documentation forms a critical component of your ISO 13485 file while supporting broader compliance objectives.
Validation documentation includes:
- Validation protocols defining test parameters and acceptance criteria
- Test results demonstrating process capability
- Statistical analyses supporting process control decisions
- Change control records when processes are modified
- Revalidation data when required by risk assessments
Medical Device Design Control File Integration
Medical device design control file integration ensures your development processes align with quality management system requirements. This integration creates seamless workflows that support both innovation and compliance objectives.
Design control integration benefits include reduced development cycle times, improved regulatory submission quality, and enhanced traceability between design requirements and manufacturing specifications.

What is ISO 13485 Quality Management System File Implementation
Implementing an ISO 13485 quality management system file structure requires careful planning and systematic execution. The process involves more than just organizing documents; it requires creating workflows that support both compliance and operational efficiency.
Successful implementation transforms documentation from a compliance burden into a strategic advantage that supports business growth and operational excellence. When done correctly, your file system becomes a competitive differentiator in regulated markets.
Medical Device Standards Alignment in File Structure
Aligning your file structure with medical device Standards ensures consistency with industry best practices while supporting regulatory requirements. This alignment creates efficiencies that reduce compliance costs while improving audit outcomes.
BPRHub automatically aligns file structures with multiple Standards, ensuring manufacturers meet requirements across different regulatory jurisdictions without duplicating efforts.
ISO 13485 Quality System Documentation Best Practices
Implementing ISO 13485 quality system documentation best practices creates sustainable compliance systems that support long-term business success. These practices emphasize efficiency, accuracy, and auditability while reducing administrative burden.
Best practices include:
- Standardized templates that ensure consistency
- Automated workflows that reduce manual errors
- Integration capabilities that eliminate duplication
- Real-time monitoring that identifies potential issues
- Continuous improvement processes that enhance effectiveness
Medical Device Risk Management in ISO 13485 Files
Integrating medical device risk management into your ISO 13485 files creates comprehensive approaches to patient safety and product quality. This integration ensures risk considerations inform all aspects of your quality management system while supporting regulatory requirements.
Risk management documentation must demonstrate systematic identification, evaluation, and control of risks throughout the product lifecycle. This documentation forms a critical component of your ISO 13485 file while supporting broader safety objectives.
Risk Assessment Documentation Requirements
Risk assessment documentation must provide clear evidence of systematic risk identification and evaluation processes. This documentation supports both ISO 13485 requirements and broader regulatory compliance objectives.
Effective risk assessment documentation includes hazard identification, risk estimation, risk evaluation, and risk control measure implementation. Each element must be traceable and maintainable throughout the product lifecycle.
Risk Control Measure File Integration
Risk control measure integration ensures your quality management system effectively addresses identified risks while maintaining operational efficiency. This integration creates accountability systems that support patient safety objectives.
Control measure documentation must demonstrate effectiveness while providing clear implementation guidance for personnel. Regular review and updating ensure continued relevance and effectiveness.
ISO 13485 Audit Preparation Using Medical Device Files
Preparing for ISO 13485 audit activities requires systematic organization of your medical device files to support efficient review processes. Well-organized files demonstrate quality management system maturity while reducing audit duration and costs.
Audit preparation involves more than just organizing documents—it requires creating narratives that clearly communicate your compliance approach and demonstrate systematic implementation of quality management principles.
Medical Device Compliance Audit Trail Documentation
Medical device compliance audit trail documentation provides clear pathways for auditors to verify implementation of your quality management system. These trails must demonstrate systematic approaches while supporting regulatory requirements.
Effective audit trails include:
- Clear linkages between policies and implementation
- Evidence of systematic review and improvement
- Traceability between requirements and evidence
- Documentation of corrective and preventive actions
- Records of management review and decision-making
ISO 13485 Guidelines for Audit-Ready Files
Following ISO 13485 guidelines for audit preparation ensures your files support efficient review processes while demonstrating compliance effectiveness. These guidelines emphasize organization, accessibility, and completeness.
Audit-ready files include clear indexing systems, comprehensive cross-references, and evidence of ongoing maintenance and review. When properly organized, these files accelerate audit processes while building auditor confidence.
ISO 13485 Certification Support Through Complete Device Files
Supporting ISO 13485 certification requires comprehensive device files that demonstrate systematic implementation of quality management principles. These files serve as primary evidence during certification audits while supporting ongoing compliance activities.
Certification preparation involves creating compelling narratives that clearly communicate your quality commitment while providing auditors with efficient access to supporting evidence.
ISO Medical Device Certification Documentation
ISO medical device certification documentation must provide comprehensive evidence of quality management system effectiveness. This documentation supports certification objectives while creating foundations for ongoing compliance.
Certification documentation includes quality manuals, procedure documents, work instructions, and records demonstrating implementation effectiveness. Each element must support the overall compliance narrative while meeting specific regulatory requirements.
Medical Device Regulation Compliance File Requirements
Medical device regulation compliance file requirements vary by jurisdiction but share common elements that support patient safety and product quality objectives. Understanding these requirements enables manufacturers to create efficient compliance systems.
BPRHub addresses these varying requirements through configurable compliance frameworks that adapt to different regulatory environments while maintaining consistency in core quality management approaches.
How BPRHub Helps with ISO 13485:2016 Practices
BPRHub transforms medical device compliance from a complex challenge into a strategic advantage through our all-in-one Quality, Compliance, and Governance. Our Unified Compliance Framework centralizes management of 30+ Standards including ISO 13485:2016, FDA, and GMP requirements within a single, integrated interface.
We digitise document analysis, automates audit workflows, and centralizes compliance data to streamline regulatory compliance while eliminating manual processes. This automation reduces errors and ensures manufacturers remain audit-ready, transforming compliance from a reactive burden into a proactive growth enabler.
BPR Hub provides real-time operational insights and vendor traceability, enabling manufacturers to identify and prioritize risks before they impact operations. With centralized Batch Manufacturing Records (BMRs) and automated compliance monitoring, BPRHub frees up resources for innovation while ensuring consistent adherence to ISO 13485 requirements.
Streamline ISO 13485 docs, book your BPRHub demo now!
📍 Book a Demo
📧 hello@bprhub.com
Key Takeaways
→ Medical device files under ISO 13485 serve as comprehensive repositories demonstrating quality management system effectiveness and regulatory compliance throughout the product lifecycle
→ Proper file organization integrates technical documentation, risk management, and quality system records to create audit-ready compliance systems that support business growth
→ BPRHub's Unified Compliance Framework centralizes 30+ Standards management, transforming documentation from administrative burden into strategic competitive advantage
→ Effective implementation requires systematic approaches to document control, change management, and audit preparation that demonstrate ongoing compliance commitment
→ Integration capabilities between design controls, manufacturing records, and quality systems create efficiencies that reduce compliance costs while improving regulatory outcomes
→ Continuous maintenanace of medical device files ensures ongoing compliance effectiveness while supporting innovation and market expansion objectives
FAQ
Q. What should a medical device file include?
A medical device file must include device specifications and intended use documentation, risk management analyses and control measures, clinical evaluation data and post-market surveillance information, manufacturing specifications and process validation records, labeling and instructions for use, and quality management system documentation demonstrating compliance with ISO 13485 standard requirements.
Q. What does ISO 13485 specify requirements for?
ISO 13485 specifies requirements for quality management systems specifically designed for medical device organizations. It establishes requirements for design and development, risk management, purchasing and supplier controls, production and service provision, measurement and improvement processes, and comprehensive documentation systems that demonstrate ongoing compliance with regulatory requirements and patient safety objectives.
Q. What is the SOP for medical device files?
Medical device file SOPs establish systematic procedures for creating, reviewing, approving, and maintaining documentation throughout the product lifecycle. These procedures include document control protocols, change management processes, version control systems, approval hierarchies, and distribution controls. The SOP ensures consistency while supporting ISO 13485 documentation requirements and regulatory compliance objectives.
Q. What is ISO 13485 standard for medical devices?
The ISO 13485 standard establishes quality management system requirements specifically for medical device manufacturers and related service organizations. It provides a framework for demonstrating ability to provide medical devices that consistently meet customer and regulatory requirements. The standard emphasizes risk-based approaches, regulatory compliance, and continuous improvement throughout the device lifecycle.
Q. What is the difference between ISO 9001 and cGMP?
ISO 9001 provides general quality management system requirements applicable across industries, while cGMP (current Good Manufacturing Practice) establishes specific manufacturing requirements for regulated products including medical devices. ISO 9001 focuses on customer satisfaction and continuous improvement, whereas cGMP emphasizes product safety, identity, strength, purity, and quality through detailed manufacturing controls and documentation requirements.
Get insights that help you minimize risks and maximize profits.
Dive deeper into manufacturing compliance with our free resources.
We get it, compliance can get tough.
Here are some additional resources to help.
We get it, compliance can get tough. Here are some additional resources to help.




Get updates in your inbox

%20(1).svg)

.avif)

