.svg)
%20(1).svg)

Medical device manufacturers face complex regulatory pathways that can make or break market entry timelines. Understanding FDA medical device classes becomes critical as improper classification can lead to significant regulatory delays, increased costs, and potential compliance issues that could have been prevented with proper regulatory planning. The medical device classification system directly impacts your time-to-market, regulatory costs, and compliance requirements.
This comprehensive guide breaks down the three FDA medical device classes, from low-risk Class I devices requiring basic general controls to high-risk Class III devices demanding extensive premarket approval processes that transform regulatory complexity into strategic advantage.
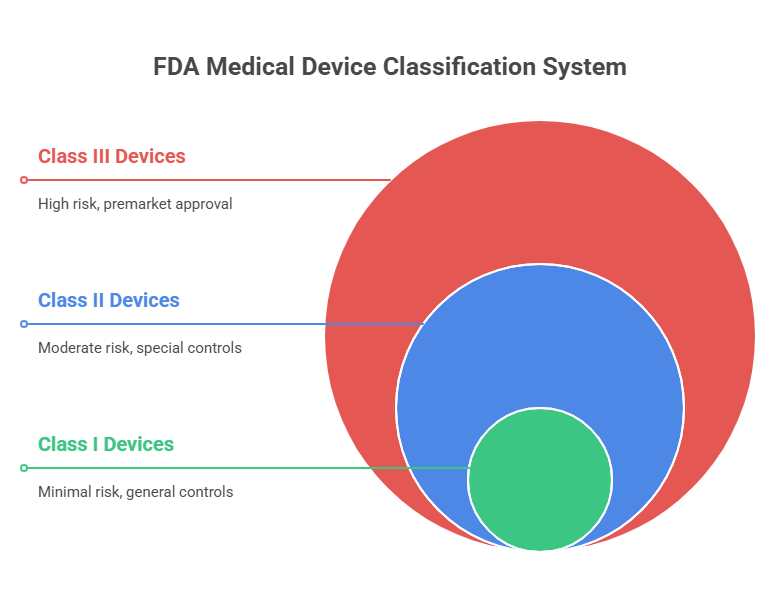
Understanding the FDA Medical Device Classification System
The medical device classification system serves as the foundation for all FDA regulatory requirements, determining the specific pathway your device must follow to reach the market. This risk-based framework ensures patient safety while providing manufacturers with clear regulatory roadmaps that balance innovation with protection.
The FDA classification system evolved from the Medical Device Amendments of 1976, creating a structured approach that categorizes devices based on their potential risk to patients and users. This systematic classification enables manufacturers to understand regulatory requirements early in development, preventing costly delays and ensuring efficient market entry.
How Medical Device Classification Works
Device classification operates through a systematic evaluation of intended use, indications for use, and the level of control necessary to provide reasonable assurance of safety and effectiveness. The FDA assigns each device to one of three classes based on the risk level associated with its use.
The classification process considers factors including the device's intended use, the degree of risk to patients, and whether existing general controls provide adequate safety assurance. Higher-risk devices require additional controls and more extensive clinical evidence to demonstrate safety and effectiveness.
Manufacturers must understand that device classification affects every aspect of regulatory strategy, from initial development planning through post-market surveillance requirements. Early classification determination enables strategic planning that aligns development activities with regulatory expectations.
Risk-Based Classification Approach for Medical Devices
The medical device classification system uses a risk-based approach that evaluates potential harm to patients if the device fails or malfunctions. This scientific methodology ensures that regulatory requirements are proportional to actual risk levels while maintaining innovation pathways for low-risk devices.
Class I devices present minimal risk and are subject to general controls, including establishment registration, device listing, and adherence to Quality System Regulation (QSR) requirements. These devices typically don't require premarket approval or clearance, enabling faster market entry for manufacturers.
The risk assessment considers both the probability of device failure and the severity of consequences if failure occurs. This dual analysis ensures that regulatory requirements appropriately address actual patient risk while avoiding unnecessary barriers for safe, effective devices.
Streamline FDA Compliance with BPRHub – Get Started Today
📍 Book a Demo
📧 hello@bprhub.com
Complete Breakdown of Class 1 Medical Device Requirements
Class 1 medical devices represent the lowest risk category within the FDA classification system, encompassing devices that present minimal potential for harm to patients. These devices are subject to general controls that provide basic safety assurance without requiring extensive premarket review processes.
The streamlined regulatory pathway for Class I devices enables manufacturers to bring safe, effective products to market more quickly while maintaining appropriate safety oversight. This efficiency benefits both manufacturers seeking market entry and patients needing access to beneficial medical technologies.
FDA Class 1 Medical Device Characteristics
FDA Class 1 medical devices share common characteristics that distinguish them from higher-risk categories. These devices typically perform simple functions, have well-understood technology, present minimal risk of injury, and don't sustain or support life-critical functions.
Key characteristics include simple design and function, minimal patient contact or interaction, well-established technology with known safety profiles, and minimal potential for harm if the device malfunctions. These factors combine to create a risk profile that general controls can adequately address.
Most Class I devices are exempt from premarket notification (510(k)) requirements, though some Class I devices specifically listed in FDA regulations still require 510(k) clearance. This exemption significantly reduces regulatory burden for the majority of Class I devices while maintaining safety oversight through general controls.
Types of Medical Devices in Class I Category
Types of medical devices in the Class I category encompass a wide range of medical specialties and applications, from basic examination tools to simple therapeutic devices. Understanding these categories helps manufacturers identify appropriate regulatory pathways for their products.
Common Class I device categories include surgical instruments like scissors and forceps, examination gloves, bandages and wound dressings, stethoscopes, and simple exercise equipment. Each category shares the common characteristic of presenting minimal risk to patient safety.
Low-Risk Class 1 Device Examples
Low-risk Class I device examples demonstrate the diversity of products that meet minimal risk criteria. Elastic bandages provide wound support without complex mechanisms, while tongue depressors enable simple diagnostic examination procedures.
Manual wheelchairs represent mechanical devices that assist patient mobility without electronic controls or complex systems. These examples demonstrate how Class I devices deliver essential healthcare functions through straightforward, well-understood mechanisms.
Additional examples include examination gloves, bed pans, and hand-held surgical instruments. Each device serves important medical functions while maintaining simple designs that minimize potential patient harm.
General Controls for Class I Devices
General controls for Class I devices establish baseline safety requirements that all medical devices must meet, regardless of classification. These controls provide fundamental protections while avoiding unnecessary regulatory burden for low-risk devices.
General controls include establishment registration and device listing with the FDA, adherence to Quality System Regulation (21 CFR Part 820), proper labeling requirements including intended use and contraindications, and medical device reporting (MDR) for adverse events. These requirements create accountability without impeding innovation. Note that the FDA is transitioning toward ISO 13485 harmonization for quality system requirements.
Manufacturers must also comply with FDA inspection requirements, maintain complaint files, and ensure that devices are manufactured by good manufacturing practices. These controls provide systematic oversight that protects patients while enabling efficient manufacturing operations.
Class 2 Medical Device Standards and Examples
Class 2 medical devices occupy the middle tier of FDA classification, representing moderate-risk devices that require general controls plus special controls to provide reasonable assurance of safety and effectiveness. These devices typically require a 510(k) premarket notification demonstrating substantial equivalence to legally marketed predicate devices.
The moderate-risk classification reflects devices that present some potential for patient harm but don't sustain or support life or present unreasonable risk of illness or injury. This balanced risk profile requires additional regulatory oversight beyond Class I requirements while avoiding the extensive approval process required for Class III devices.
FDA Class 2 Medical Device Requirements
FDA Class 2 medical device requirements build upon general controls by adding special controls specific to device types or categories. These additional requirements address unique risks associated with particular device functions or patient populations.
Special controls may include performance standards, postmarket surveillance requirements, patient registries, special labeling requirements, premarket data requirements, or guidelines for clinical evaluation. These controls are developed based on scientific understanding of device-specific risks and mitigation strategies.
Most Class II devices require a 510(k) premarket notification unless specifically exempt. The 510(k) process requires manufacturers to demonstrate that their device is substantially equivalent to a legally marketed predicate device, providing a streamlined approval pathway compared to premarket approval (PMA) requirements.
510(k) FDA Clearance Process for Class II Devices
The FDA clearance process through 510(k) premarket notification provides an efficient pathway for Class II devices to reach the market while maintaining appropriate safety oversight. This process focuses on demonstrating substantial equivalence rather than absolute safety and effectiveness.
Substantial equivalence means the new device has the same intended use as a predicate device and either has the same technological characteristics or has different technological characteristics that don't raise new questions of safety and effectiveness. This standard enables innovation while maintaining safety assurance.
The 510(k) review process typically takes 90 days, though complex devices may require additional time for FDA review. Manufacturers can accelerate the process by conducting thorough predicate research, preparing comprehensive submissions, and engaging in pre-submission meetings with FDA reviewers.
Moderate-Risk Class II Device Examples
Moderate-risk Class II device examples demonstrate the diversity of products requiring balanced regulatory oversight. Powered wheelchairs include electrical systems that require additional safety controls compared to manual alternatives.
Infusion pumps deliver medications directly to patients, requiring precise dosing controls and safety mechanisms to prevent overdosing or underdosing. These devices illustrate how moderate complexity increases regulatory requirements.
Pregnancy test kits represent diagnostic devices that influence medical decision-making, requiring accuracy standards and appropriate labeling to ensure proper use and interpretation of results.
Special Controls Beyond General Requirements
Special controls beyond general requirements address device-specific risks that general controls cannot adequately address. These controls are developed through scientific evaluation of device performance, clinical use patterns, and potential failure modes.
Examples of special controls include performance standards specifying minimum accuracy requirements for diagnostic devices, guidance documents providing manufacturers with FDA expectations for specific device types, and postmarket surveillance requirements for devices with evolving safety profiles.
The FDA develops special controls through collaborative processes involving manufacturers, healthcare providers, and patient advocates. This stakeholder input ensures that special controls address real-world risks while maintaining practical feasibility for manufacturers.
Class 3 Medical Device Approval Process
Class 3 medical devices represent the highest-risk category within FDA classification, encompassing devices that sustain or support life, are implanted, or present an unreasonable risk of illness or injury. These devices require the most stringent regulatory oversight through premarket approval (PMA) processes.
The high-risk classification reflects the potential for serious patient harm if these devices fail or malfunction. This risk profile necessitates extensive clinical evidence demonstrating safety and effectiveness before market approval, ensuring that benefits outweigh risks for intended patient populations.
High-Risk FDA Class 3 Medical Device Criteria
FDA Class 3 medical device criteria focus on devices that sustain or support life, are implanted in the body, present a potential unreasonable risk of illness or injury, or are purported or represented to be for use in supporting or sustaining human life. These criteria reflect the highest level of patient risk requiring comprehensive regulatory oversight.
Devices meeting Class III criteria include heart valves, pacemakers, breast implants, and life-support ventilators. Each device type presents unique risks that require extensive clinical evaluation and ongoing post-market surveillance to ensure continued safety and effectiveness.
The FDA applies strict scrutiny to Class III devices because failure can result in death or serious injury. This scrutiny requires manufacturers to invest significantly in clinical research, quality systems, and post-market monitoring to demonstrate and maintain device safety.
FDA Medical Device Approval Requirements for Class III
FDA medical device approval requirements for Class III devices center on premarket approval (PMA), the most stringent regulatory pathway requiring extensive clinical data demonstrating safety and effectiveness. This process ensures that high-risk devices meet the highest standards before reaching patients.
PMA applications must include comprehensive clinical studies demonstrating that device benefits outweigh risks for intended patient populations. These studies typically involve randomized controlled trials comparing the investigational device to the standard of care or control groups.
The PMA review process can take 6-12 months or longer, depending on device complexity and clinical data quality. FDA review includes evaluation of clinical data, manufacturing quality systems, labeling adequacy, and risk management strategies.
Life-Sustaining Class III Device Examples
Life-sustaining Class III device examples demonstrate the critical nature of devices requiring premarket approval. Ventricular assist devices support heart function in patients with severe heart failure, directly sustaining life through mechanical circulatory support.
Implantable cardioverter defibrillators (ICDs) monitor heart rhythm and deliver life-saving electrical therapy when dangerous arrhythmias occur. These devices represent the intersection of life-sustaining function and implantable technology.
Heart valve replacements replace diseased natural valves, directly impacting cardiac function and patient survival. The permanent implantation and critical function require extensive clinical evidence and ongoing surveillance.
Premarket Approval (PMA) Process
The premarket approval (PMA) process represents the FDA's most rigorous device evaluation pathway, requiring comprehensive evidence that device benefits outweigh risks for intended patient populations. This process ensures that life-sustaining and high-risk devices meet the highest safety and effectiveness standards.
PMA submissions must include complete device descriptions, intended use statements, comprehensive clinical data from well-controlled studies, detailed manufacturing information, proposed labeling, and risk analysis and management strategies. This comprehensive submission enables a thorough FDA evaluation.
The PMA review process includes scientific review by FDA medical officers, statistical analysis of clinical data, quality system inspection of manufacturing facilities, and advisory panel meetings for complex or controversial devices. This multi-faceted review ensures a comprehensive evaluation.

Key Differences Between FDA Medical Device Classes
Understanding the key differences between FDA medical device classification categories enables manufacturers to plan appropriate regulatory strategies, allocate resources effectively, and set realistic market entry timelines. These differences affect every aspect of device development and commercialization.
The primary differences center on risk level, regulatory requirements, approval pathways, and time to market. Each classification tier builds upon previous requirements while adding additional controls proportional to device risk.
Regulatory Requirements Comparison Across Classes
FDA requirements for medical devices vary significantly across classification levels, creating distinct regulatory pathways that manufacturers must navigate. Understanding these differences enables strategic planning that aligns development activities with regulatory expectations.
Class I devices require general controls, including establishment registration, device listing, QSR compliance, and appropriate labeling. Most Class I devices are exempt from premarket review, enabling direct market entry after meeting general control requirements.
Class II devices require general controls plus special controls and typically require a 510(k) premarket notification demonstrating substantial equivalence. Special controls address device-specific risks through performance standards, guidance documents, or postmarket requirements.
Class III devices require general controls, special controls where applicable, and premarket approval (PMA) demonstrating safety and effectiveness through clinical data. This comprehensive approach reflects the high-risk nature of Class III devices.
Risk Levels and Control Measures by Class
Risk levels and control measures increase progressively from Class I through Class III, reflecting the FDA's risk-based regulatory approach. This systematic escalation ensures that regulatory requirements are proportional to actual patient risk.
Class I devices present minimal risk with control measures focused on basic manufacturing quality and appropriate labeling. Class II devices present a moderate risk requiring additional controls addressing specific device risks. Class III devices present a high risk, requiring comprehensive clinical evidence and ongoing surveillance.
General Controls vs Special Controls vs PMA
General controls provide baseline requirements for all medical devices, including registration, listing, QSR compliance, labeling, and medical device reporting. These fundamental requirements ensure basic manufacturing quality and appropriate device identification.
Special controls address device-specific risks that general controls cannot adequately manage. These may include performance standards, clinical evaluation guidance, postmarket surveillance requirements, or patient registries tailored to specific device types.
PMA requirements demand comprehensive clinical evidence demonstrating that device benefits outweigh risks for intended patient populations. This evidence includes controlled clinical studies, detailed risk analysis, and comprehensive quality system documentation.
Time to Market Differences
Time to market differences across device classes reflect varying regulatory complexity and evidence requirements. Class I devices are exempt from premarket review and can reach the market immediately after meeting general control requirements.
Class II devices requiring 510(k) clearance typically take 3-6 months from submission to clearance, assuming a successful initial review. Additional time may be required for predicate research, submission preparation, and potential FDA questions.
Class III devices requiring PMA approval typically take 12-18 months or longer from submission to approval. This timeline includes extensive clinical data generation, comprehensive submission preparation, and thorough FDA review processes.
Medical Devices Examples by Classification Category
Medical device examples across classification categories demonstrate the practical application of FDA risk-based classification principles. Understanding real-world device examples helps manufacturers identify appropriate regulatory pathways and plan development strategies accordingly.
Each classification category encompasses diverse device types serving different medical needs while sharing common risk characteristics that determine regulatory requirements.
FDA Approved Devices in Each Class
FDA-approved devices represent successful navigation of regulatory pathways appropriate to their risk classification. These examples provide practical illustrations of how different device types align with FDA classification principles.
Class I approved devices include surgical scissors, examination gloves, and elastic bandages that provide essential medical functions through simple, well-understood mechanisms requiring minimal regulatory oversight.
Class II cleared devices include blood pressure monitors, pregnancy test kits, and infusion pumps that require moderate regulatory oversight to address specific risks while enabling reasonable market access.
Class III approved devices include pacemakers, heart valves, and breast implants that require extensive clinical evidence and ongoing surveillance due to their high-risk profiles and critical patient impact.
Common Medical Device Industry Classifications
Medical device industry classifications reflect practical groupings that help manufacturers understand regulatory expectations for specific device categories. These industry-recognized classifications align with FDA regulatory frameworks while providing practical development guidance.
Diagnostic devices span all three classes depending on risk level, from simple Class I examination tools to complex Class III life-supporting diagnostic equipment. Therapeutic devices similarly span classifications based on patient risk and device complexity.
Surgical devices range from Class I handheld instruments to Class III implantable devices, with classification determined by patient risk, complexity of use, and potential for harm. This diversity illustrates how intended use and risk profile determine regulatory requirements.
How to Classify Medical Devices Using FDA Guidelines
Understanding how to classify medical devices using the FDA classification for medical devices guidelines enables manufacturers to determine appropriate regulatory pathways early in development. This early classification determination prevents costly development delays and ensures strategic regulatory planning.
The classification process involves a systematic evaluation of intended use, technological characteristics, and risk assessment using established FDA criteria and precedent decisions.
FDA Device Classification Database Search
The FDA Device Classification Database provides manufacturers with searchable access to classification decisions for thousands of medical devices. This database enables manufacturers to research similar devices and understand regulatory precedents relevant to their products.
The database includes device names, classification codes, product codes, and regulatory pathways for devices across all three classification categories. Manufacturers can search by device name, intended use, or product code to identify relevant precedents.
Effective database searching requires understanding device terminology, intended use descriptions, and classification rationale. This research forms the foundation for classification determination and regulatory strategy development.
Medical Device Compliance Steps by Class
Medical device compliance steps vary by classification, requiring manufacturers to understand class-specific requirements and develop appropriate compliance strategies. Early compliance planning prevents delays and ensures efficient regulatory navigation.
Class I compliance focuses on general controls, including establishment registration, device listing, QSR implementation, and appropriate labeling. These fundamental requirements provide baseline compliance for all medical devices.
Class II compliance adds special controls and typically requires 510(k) preparation and submission. Manufacturers must identify applicable special controls and demonstrate substantial equivalence to appropriate predicate devices.
Class III compliance requires comprehensive PMA preparation, including clinical studies, detailed risk analysis, and extensive quality system documentation. This comprehensive approach addresses the high-risk nature of Class III devices.
FDA Guidance Documents for Device Classification
FDA guidance documents provide manufacturers with detailed information about classification criteria, regulatory pathways, and submission requirements. These documents represent FDA thinking on specific device types and regulatory approaches.
Guidance documents include device-specific guidance addressing particular device categories, general guidance covering broad regulatory topics, and draft guidance representing the FDA's current thinking on emerging issues.
Manufacturers should regularly review relevant guidance documents to understand FDA expectations and incorporate current regulatory thinking into development and submission strategies. This proactive approach prevents submission delays and regulatory challenges.
The FDA regularly updates guidance documents to reflect scientific advances, regulatory experience, and stakeholder feedback. Staying current with guidance updates ensures that regulatory strategies remain aligned with FDA expectations.
How BPRHub Helps with Medical Device Compliance
BPRHub transforms medical device compliance from a complex regulatory burden into a streamlined competitive advantage through its comprehensive Quality, Compliance, and Governance platform. The system centralizes all FDA regulatory requirements across device classifications, eliminating duplicate workflows while ensuring continuous audit readiness.
With BPRHub's integrated document management and quality systems, medical device manufacturers maintain complete traceability from design controls through post-market surveillance. The platform's automated workflows ensure that ISO 13485 traceability requirements align seamlessly with FDA classification requirements, reducing regulatory complexity while accelerating time-to-market.
BPRHub's risk management modules specifically address the varying requirements across device classes, from Class I general controls to Class III PMA submissions. The platform's intelligent automation captures quality data in real-time, generates regulatory reports automatically, and provides predictive analytics that identify potential compliance issues before they impact regulatory submissions.
The system's integration with existing manufacturing infrastructure supports the quality management system requirements essential for medical device manufacturing while maintaining FDA compliance throughout the product lifecycle.
Accelerate Your Device Approval Process – Power It with BPRHub today
📍 Book a Demo
📧 hello@bprhub.com
Key Takeaways
→ FDA medical device classes are determined by risk level, with Class I presenting minimal risk, Class II moderate risk, and Class III high risk requiring progressively stringent regulatory controls
→ Medical device classification directly impacts regulatory pathway, time to market, and compliance requirements, making early classification determination critical for strategic planning
→ Class 1 medical devices require only general controls and are typically exempt from premarket review, enabling faster market entry for low-risk devices
→ Class 2 medical devices require a 510(k) premarket notification demonstrating substantial equivalence to predicate devices, balancing safety oversight with reasonable market access
→ Class 3 medical devices demand premarket approval (PMA) with comprehensive clinical evidence, reflecting the high-risk nature of life-sustaining and implantable devices
→ BPRHub's compliance platform centralizes medical device regulatory requirements across all classifications, automating workflows and ensuring continuous audit readiness while accelerating regulatory submissions
FAQ
Q. What are Class 1, 2, and 3 device examples?
Class 1 devices include elastic bandages, examination gloves, and manual wheelchairs that present minimal patient risk. Class 2 devices include blood pressure monitors, infusion pumps, and pregnancy test kits requiring moderate regulatory oversight. Class 3 devices include pacemakers, heart valves, and ventricular assist devices that sustain life or present high risk, requiring extensive clinical evidence through premarket approval processes.
Q. What is the difference between a class 1 and a class 2 medical device?
Class 1 medical devices require only general controls and are typically exempt from premarket review, while class 2 medical devices require general controls plus special controls and usually need 510(k) premarket clearance. Class 1 devices present minimal risk and can reach the market immediately after meeting basic requirements, whereas Class 2 devices present moderate risk, requiring substantial equivalence demonstration to predicate devices before market entry.
Q. What is class 1 IIa, IIb, or iii?
The Class I, IIa, IIb, and III system refers to European Union medical device classification under the Medical Device Regulation (MDR), which differs from the FDA medical device classification system. The FDA uses Class I, II, and III based on risk levels, while the EU system includes four classes, with IIa and IIb representing different moderate-risk categories. Understanding both systems is essential for manufacturers planning global market strategies.
Q. What is an example of a Class 1 device?
A Class 1 device example is an elastic bandage, which provides wound support through simple mechanical compression without complex mechanisms or significant patient risk. Other examples include examination gloves that provide barrier protection, tongue depressors for diagnostic examination, and manual wheelchairs for patient mobility. These devices share characteristics of simple function, minimal patient risk, and well-understood technology requiring only general regulatory controls.
Get insights that help you minimize risks and maximize profits.
Dive deeper into manufacturing compliance with our free resources.
We get it, compliance can get tough.
Here are some additional resources to help.
We get it, compliance can get tough. Here are some additional resources to help.




Get updates in your inbox

%20(1).svg)

.avif)

